아데노 부속 바이러스(Adeno-Associated Virus, AAV)는 현대 유전자 치료에서 가장 중요한 전달 플랫폼 중 하나로 자리 잡았습니다. 많은 희귀 유전 질환, 특히 기능 상실 돌연변이(loss-of-function mutations)로 인해 발생하는 질환의 경우, AAV 매개 유전자 대체 요법은 단 한 번 또는 드문 투여만으로도 지속적인 치료 효과를 제공할 가능성이 있습니다. 그러나 AAV 프로그램을 동물 연구에서 최초 인체 대상 임상시험(first-in-human clinical testing) 단계로 이행할 때는 핵심적인 전이(translational) 질문에 직면하게 됩니다. “초기 인간 투여량을 어떻게 설정하고 그 정당성을 입증할 것인가?”
흔히 범하는 실수는 마우스 투여량을 동일한 체중당 벡터 게놈(vector genomes per kilogram, vg/kg) 값을 기준으로 삼고, 여기에 일반적인 안전 계수(safety factor)를 곱해 직접 인간 투여량으로 변환하는 것입니다. 이러한 접근 방식은 오해를 불러일으킬 수 있습니다. AAV의 용량-반응(dose-response) 관계는 표적 장기의 크기, 종 특이적 형질도입(transduction), 캡시드 생물학, 면역 인지, 생체 내 분포(biodistribution), 제품 품질, 효능(potency) 및 투여 경로에 의해 결정되기 때문입니다. 그 결과, 명목상 동일한 vg/kg 용량을 투여받은 두 종이라 할지라도 생물학적 노출과 안전성 프로파일은 크게 다르게 나타날 수 있습니다.
따라서 임상 1상 초기 투여량에 대한 합리적인 근거 설정은 단순히 수학적 계산 공식 하나로 해결되지 않습니다. 이는 효능, 생체 내 분포, 독성, 역가(potency), 제조 동등성, 투여 경로별 제약 조건, 환자의 위험도 및 규제 전략을 종합적으로 통합하는 구조화된 과학적 논증이어야 합니다.
- 마우스 데이터는 개념 검증(Proof of Concept)에는 유용하지만, 초기 인간 투여량을 결정할 때 단독으로 사용되어서는 안 됩니다.
- 전신 투여(Systemic) AAV의 경우, 비인류 영장류(NHP) 또는 기타 관련 대동물 데이터가 생체 내 분포 및 안전성에 관한 결정적인 정보를 제공하는 경우가 많습니다.
- 안구(ocular) 또는 중추신경계(CNS) 투여와 같은 국소 전달(local delivery)의 경우, vg/kg 보다는 안구당 벡터 게놈 수(vg/eye), 주사 부위당 벡터 게놈 수(vg/injection site) 또는 해당 구획 내 총 벡터 게놈 수(total vg)가 더 유의미한 투여량 단위일 수 있습니다.
- 규제 기관들은 일반적으로 고정된 환산 공식보다는 제품의 특성을 반영하고 과학적으로 입증된 투여량 근거(dose rationale)를 요구합니다.
- CMC(화학·제조·품질관리) 및 분석적 동등성은 투여량 환산의 핵심입니다. 명목상의 vg 투여량은 벡터의 역가와 품질이 명확히 규명되었을 때만 의미를 갖기 때문입니다.
1. AAV 투여량 환산이 단순히 vg/kg 계산을 넘어서는 이유
AAV 투여량은 일반적으로 체중 1킬로그램당 벡터 게놈 수(vg/kg)로 보고됩니다. 이 단위는 체중에 맞추어 용량을 표준화하고 동물과 환자 간의 비교를 가능하게 하므로 전신 투여 시 유용합니다. 반면, 국소 투여에서는 안구당, 주사 부위당, 뇌 영역당 또는 투여 구획당 총 벡터 게놈 수로 투여량을 보고하기도 합니다.
문제는 vg/kg 단위 자체가 아니라, 마우스에서 인간으로 직접 환산할 때 깔려 있는 암묵적인 가정입니다. 즉, “체중 1kg이 종 전반에 걸쳐 벡터 분포, 표적 장기 노출, 면역 활성화 및 치료 반응과 항상 동일한 관계를 갖는다”는 가정입니다. AAV의 경우, 이 가정은 대개 틀린 경우가 많습니다.
AAV 벡터는 생물학적 전달 시스템입니다. 체내를 순환하고, 분포하며, 세포 표면의 글리칸(당쇄)이나 수용체에 결합하고, 세포 내로 들어가 핵으로 벡터 게놈을 전달해 이식유전자(transgene) 발현을 유도합니다. 동시에 선천성 및 적응성 면역 반응을 유발할 수도 있습니다. 따라서 AAV 투여량을 설정할 때는 약물학적 노출과 생물학적 활성을 모두 고려해야 합니다.
최근의 규제 기대치도 이러한 복잡성을 반영하고 있습니다. 희귀 질환 유전자 치료제에 대한 FDA 가이드라인에 따르면, 전임상 단계에서 생물학적으로 활성이 있는 용량 범위를 확인하고, 초기 임상 용량 및 증량 계획을 추천하며, 임상 투여 경로의 타당성과 합리적인 안전성을 입증하고, 독성 및 모니터링 변수를 규명할 수 있도록 지원해야 한다고 명시하고 있습니다. EMA 가이드라인 역시 투여량 선택이 제품의 역가(potency)와 연계되어야 하며, 품질 및 비임상 데이터에 의해 뒷받침되어야 함을 강조하고 있습니다.
2. 단순한 마우스-인간 AAV 스케일링이 왜 위험할 수 있는가?
마우스 모델에서 인간으로 전환할 때, 여러 생물학적 및 기술적 요인들이 AAV의 실질적인 유효 용량을 체계적으로 변화시킬 수 있습니다.
- 표적 장기의 크기와 세포 수는 체중에 비례하지 않습니다.
예를 들어 간 표적 AAV 치료에서 간이 전체 체중에서 차지하는 비율은 마우스, 비인류 영장류, 영유아, 성인마다 다릅니다. 동일한 vg/kg 용량을 투여하더라도 간세포(hepatocyte)당 이용 가능한 벡터 입자의 수는 크게 다를 수 있습니다. 망막, 근육, 중추신경계 영역 및 기타 표적 조직에도 동일한 원리가 적용됩니다. - AAV 수용체 생물학 및 글리칸 발현 양상이 종마다 다릅니다.
다양한 AAV 혈청형(serotypes)은 서로 다른 세포 부착 인자와 세포 진입 경로에 의존합니다. 예를 들어 AAV9은 단일 범용 수용체가 아니라 말단 갈락토스 함유 글리칸의 영향을 받습니다. 글리칸의 풍부함, 접근성 및 조직 분포의 차이로 인해 설치류에서의 형질도입 효율만 가지고는 인간에서의 형질도입 결과를 예측하기 어렵습니다. - 인간의 면역 생물학은 무균(SPF) 마우스 모델에서 완벽히 재현되지 않습니다.
임상시험 환자들은 기존에 형성된 항 AAV 항체(pre-existing anti-AAV antibodies), 기억 T 세포 반응,补체 활성화 위험, 기저 염증 또는 질환과 관련된 장기 취약성을 가질 수 있습니다. 중화항체 분석법에서 음성으로 판명된 환자라 할지라도, 선천성 면역 활성화 및 항-캡시드 T 세포 반응으로 인해 전달 효율과 안전성이 달라질 수 있습니다. - 제품 품질이 투여량의 실질적인 의미를 좌우합니다.
동일한 게놈 역가를 가진 두 AAV 배치(lot)라 하더라도 빈/가득 찬 캡시드 비율(empty/full capsid ratio), 게놈 무결성(genome integrity), 역가, 응집(aggregation), 잔류 DNA, 잔류 단백질, 감염성 및 전반적인 생물학적 활성 면에서 차이가 날 수 있습니다. qPCR 또는 ddPCR로 측정한 게놈 역가는 기능적 역가 측정치와 완전히 동일하지 않습니다. 따라서 초기 논문에서 vg/동물 단위로 보고된 투여량은 임상 등급 배치와 직접 비교하기 어려울 수 있습니다. - 알로메트릭 스케일링(Allometric scaling)은 보조적 참고 자료일 뿐, 만능 해결책이 아닙니다.
전형적인 알로메트릭 스케일링(동물 간 체중 차이에 따른 생리학적 대사율 등을 고려한 환산 공식)은 많은 생리학적 과정이 체중과 비선형적 관계를 보이기 때문에 일반 제약학에서 널리 쓰입니다. 그러나 AAV는 체내에 분포하고, 결합하며, 세포에 진입하고, 유전물질을 발현시키고, 면역 반응을 촉발하는 ‘생물학적 입자’처럼 작용합니다. 따라서 알로메트릭 스케일링은 AAV 고유의 생체 내 분포, 약리학, 독성 및 역가 데이터를 대체하는 것이 아니라 보조하는 용도로만 사용되어야 합니다.
3. AAV 투여량 환산의 4가지 주요 접근법
모든 AAV 프로그램에 일률적으로 적용할 수 있는 단일 방법론은 존재하지 않습니다. 가장 적합한 투여량 환산 전략은 투여 경로, 표적 조직, 질환 생물학, 캡시드, 벡터 게놈, 치료 영역(therapeutic window), 환자군, 그리고 가용한 전임상 데이터에 따라 달라집니다.
표 1. 주요 AAV 투여량 환산 방식 및 적용 사례
| 투여량 환산 접근법 | 핵심 논리 (Core logic) | 최적의 적용 사례 (Best-fit use cases) | 주요 한계점 (Key limitations) |
| 체중 기준 스케일링 (Body-weight scaling) | 동물 투여량을 직접 인간의 vg/kg으로 변환하며, 보통 안전 계수를 함께 고려함. | 초기 내부 평가 단계, 혹은 뚜렷하게 지배적인 표적 장기가 없는 전신 투여 프로그램. | 생체 내 분포, 표적 장기 크기, 형질도입 효율 및 면역 반응의 종간 차이를 지나치게 단순화함. |
| 체표면적 또는 알로메트릭 스케일링 | 체중 또는 체표면적을 기반으로 한 비선형 스케일링 공식을 적용함. | 전신 노출 평가나 독성 평가 시 보조적인 계산법으로 활용됨. | AAV는 전통적인 소분자 화합물이 아니므로, 캡시드 생물학, 수용체 결합, 조직 지향성으로 인해 직접 적용에 한계가 있음. |
| 표적 장기 기준 스케일링 (Target-organ scaling) | 표적 장기의 질량, 세포 수 또는 해부학적 구획을 기반으로 용량을 조정함. | 간 표적, 안구, 중추신경계(CNS) 및 특정 장기 중심의 AAV 프로그램. | 신뢰할 수 있는 장기 크기, 세포 수 및 분포 데이터가 필요하며, 면역 반응이나 표적 외 효과(off-target effects)를 놓칠 수 있음. |
| 비인류 영장류(NHP) 기반 스케일링 | 비인류 영장류에서의 효능, 생체 내 분포 및 독성 데이터를 전환의 기준(Anchor)으로 삼음. | 고용량 전신 투여 AAV 프로그램 및 임상시험계획서(IND) 제출을 위한 용량 정당성 입증 단계. | NHP 연구는 비용이 많이 들고 표본 수가 적은 경우가 많으며, 여전히 인간의 반응을 완벽하게 예측하지는 못함. |
실무적으로 대부분의 임상 단계 AAV 프로그램은 이 방법들을 혼합하여 사용합니다. 체중 기준 계산으로 대략적인 기준점을 잡고, 표적 장기 스케일링을 통해 예상 노출량을 정밀화하며, NHP 데이터를 바탕으로 안전성 한계(safety margins)와 중점 모니터링 대상을 설정합니다. 최종적인 임상 투여량 설정 근거서에는 각 방법론에 깔린 가정이 명확히 밝혀져 있어야 합니다.
4. 투여 경로에 따른 AAV 용량 스케일링 논리의 변화
AAV 투여량 환산은 전달 경로를 정의하는 것부터 시작되어야 합니다. 동일한 총 벡터 투여량이라도 정맥 주사, 척수강 내 주사, 대조조 내 주사, 망막하 주사, 유리체 내 주사, 근육 주사 또는 특정 조직 구획에의 직접 투여 여부에 따라 완전히 다른 임상적 의미를 갖습니다.
전신 정맥 투여(systemic intravenous) AAV의 경우, 투여되는 총 벡터 양(vector burden) 자체가 중요한 안전성 고려 사항입니다. 많은 AAV 혈청형은 최종 표적이 근육이나 중추신경계라 할지라도 전신 투여 후 상당량이 간에 분포하게 됩니다. 반면 국소 투여(local delivery)에서는 체중보다는 국소 해부학적 구조, 주입 부피, 조직 접근성, 표적 세포 수 및 국소 염증 위험이 더 중요합니다.
표 2. 투여 경로에 따른 AAV 용량 스케일링 분석
| 투여 경로 및 적응증 유형 | 권장 투여량 단위 | 주요 스케일링 고려사항 | 실무 권장 사항 |
| 전신 정맥 투여 AAV | vg/kg 및 총 vg (total vg) | 총 벡터 축적량, 간 노출, 면역 활성화, 체중 및 총 캡시드 탑재량. | 마우스 데이터를 초기 참고치로 삼되, 생체 내 분포 및 관련 대동물 독성 데이터를 바탕으로 임상 용량의 안전성을 확보하십시오. |
| 간 표적 AAV | vg/kg, 총 vg, 예상 간세포당 vg 수 | 간 질량, 간세포 수, 형질도입 효율, 분비 단백질의 표적 농도, 간 안전성 바이오마커. | 가능할 때마다 표적 장기 스케일링 모델과 NHP 또는 타 대동물 데이터를 결합하여 검토하십시오. |
| 안구 투여 AAV | vg/안구(vg/eye) 또는 vg/주사액 | 주입 부피, 망막 표적 세포 수, 국소 염증 반응, 전달 경로 및 벡터의 확산 범위. | vg/kg을 주요 논리로 사용하지 마십시오. 유사한 안구 AAV 프로그램 및 투여 경로를 벤치마킹하여 설정하십시오. |
| 중추신경계(CNS) 투여 AAV | vg/주사 부위, vg/뇌척수액 구획, 또는 총 vg | 뇌척수액(CSF) 부피, 국소 확산 정도, 표적 영역 커버리지, 후근신경절(DRG) 노출 및 경로 특이적 해부학 구조. | 체중 단독 기준이 아닌 해부학적 분포 영역과 안전성 확보 한계(safety margins)를 기준으로 투여량을 정하십시오. |
| 신경근육계 전신 투여 AAV | vg/kg 및 총 vg | 근육량, 간 흡수율, 면역 활성화, 환자 연령, 질병 진행 단계 및 총 벡터 축적량. | 보수적인 용량 증량 단계 설계, 강력한 안전성 모니터링 및 관련 대동물 데이터 확보가 필수적입니다. |
5. AAV 최초 인체 투여(FIH) 용량 설정을 위한 실무 가이드라인
신뢰할 수 있는 AAV 최초 인체 투여(FIH) 용량 타당성 보고서는 여러 증거 단계가 겹겹이 쌓인 종합 패키지 형태로 구축되어야 합니다. 각 단계는 제안된 임상 투여량이 질환 생물학, 벡터의 거동, 투여 경로 및 제품의 품질과 어떻게 연결되는지 설명해야 합니다.
1단계: 치료 표적 및 필요한 생물학적 효과 정의
첫 번째 질문은 “마우스에서 효과가 있었던 용량이 얼마인가?”가 아니라 “인간에게 필요한 생물학적 효과가 무엇인가?”가 되어야 합니다.
- 분비 단백질 치료제: 질병 경감 역치 이상의 혈중 단백질 농도 도달 목표 설정
- 중추신경계(CNS) 치료제: 특정 신경세포 또는 성상세포(glial) 군집으로의 형질도입
- 망막 치료제: 광수용체나 망막색소상피층에서의 충분한 발현
- 근육 질환 치료제: 기능적 변화를 이끌어내기에 충분한 근육 섬유 내 이식유전자 발현
추적해야 할 주요 데이터:
- 이식유전자 단백질 농도 또는 효소 활성도
- 표적 세포 또는 표적 조직당 벡터 게놈 복제수(copy number)
- 조직, 혈액, CSF 또는 안구액에서의 바이오마커 정상화 수준
- 질환 관련 모델에서의 기능적 회복(rescue) 지표
- 표적 결합을 증명하는 조직학적, 영상학적 또는 전기생리학적 증거
2단계: 최소 유효 용량(MED) 범위 파악
최소 유효 용량은 관련 in vitro, ex vivo, in vivo 연구를 통해 뒷받침되어야 합니다. 마우스 모델이 질환 기전을 반영한다 하더라도 마우스에서의 효과적인 단일 투여량만으로는 불충분합니다. 가능하면 용량-반응 관계(dose-response relationship) 조사를 통해 무효과 용량(no-effect dose), 최소 활성 용량, 약동학적 활성 용량, 정체(plateau) 용량(존재 시), 독성 또는 불내성 용량(toxic/poorly tolerated dose)을 모두 확보해야 합니다.
3단계: 생체 내 분포 및 발현 데이터 확보
생체 내 분포(biodistribution) 데이터는 AAV 투여량 환산의 근간입니다. 이를 통해 벡터가 어디로 이동하고, 얼마나 오래 지속되며, 어떤 조직에서 이식유전자를 발현하는지, 그리고 표적 외 조직 중 어떤 곳이 잠재적 위험에 노출되는지 파악할 수 있습니다. 전신 투여 시에는 간, 비장, 심장, 생식선, 후근신경절(DRG) 및 관련 면역 조직을 포함해야 하며, 국소 투여 시에는 주입 구획 및 인접 조직을 꼼꼼히 관찰해야 합니다.
4단계: 독성 프로파일 및 안전성 한계(Safety Margin) 설정
독성 연구는 제안된 임상 투여 경로, 용량 범위, 투여 일정 및 모니터링 계획을 최대한 사실적으로 모사해야 합니다. AAV의 안전성 평가 시에는 특히 간 손상,補체 활성화, 혈소판 감소증, 후근신경절(DRG) 병변, 염증성 사이토카인, 이식유전자 관련 독성, 벡터 유출(shedding) 및 생식선 분포 위험을 면밀히 모니터링해야 합니다.
안전 계수(safety factor)를 적용할 수 있으나 모든 AAV 프로그램에 똑같이 적용되는 만능 수치는 없습니다. 적절한 안전 한계(margin)는 질환의 중증도, 대체 치료제의 유무, 환자 연령, 투여 경로, 용량 제한 독성(DLT), 면역 위험성, 치료 영역 및 비임상 패키지의 신뢰 수준에 따라 달라집니다.
5단계: 제품 품질 및 역가 통합
벡터 제품의 특성이 정밀하게 분석되어 명확해졌을 때만 투여량 환산이 의미가 있습니다. 게놈 복제수를 기반으로 임상 용량을 설정할 때는 역가(potency), 빈/가득 찬 비율, 게놈 무결성, 잔류 숙주 세포 DNA, 잔류 플라스미드 DNA, 단백질 불순물, 응집도, 엔도톡신, 무균성, 복제 가능 AAV 테스트 및 전임상용-임상용 배치(lot) 간 동등성 평가 결과가 함께 검토되어야 합니다.
6단계: 용량 증량 및 투여 중단 규칙 설계
임상 안전성은 초기 투여량뿐만 아니라 ‘증량 설계(escalation design)’에 의해서도 좌우됩니다. 고위험군 전신 투여 AAV 프로그램은 센티넬 투여(sentinel dosing, 첫 환자 관찰 후 후속 투여), 시차 등록(staggered enrollment), 사전에 정의된 투여 중단 규칙(stopping rules), 집중적인 실험실 검사 모니터링, 면역억제 프로토콜 수립 및 독립적 데이터안전성모니터링위원회(DSMB) 운영을 포함할 수 있습니다. 지연성 면역 매개 독성은 투여 후 몇 주가 지나서 나타날 수 있으므로, 관찰 기간은 벡터 및 이식유전자의 기대 생물학적 특성에 맞춰 길게 설정해야 합니다.
표 3. AAV 최초 인체 임상(FIH) 계획을 위한 핵심 질문 리스트
| 개발 핵심 질문 | 이것이 중요한 이유 | 필요한 데이터 패키지 |
| 인간에게 필요한 생물학적 효과는 무엇인가? | 필요한 최소한의 치료적 노출 수준을 규명해 줍니다. | 표적 단백질 수준, 효소 활성, 표적 세포당 벡터 복제수, 바이오마커 반응 및 기능 회복 데이터. |
| 관련 모델에서의 최소 유효 용량은 얼마인가? | 너무 높거나 낮은 용량으로 임상을 시작하는 오류를 방지합니다. | 용량-반응 연구, 질환 모델에서의 효능 및 약동학(PD) 마커 데이터. |
| 투여 후 벡터가 체내 어디로 가며 어떻게 거동하는가? | 표적 및 표적 외 조직의 노출 위험도를 파악합니다. | 생체 내 분포(biodistribution), 체내 잔류성(persistence), 클리어런스(clearance) 및 이식유전자 발현 데이터. |
| 임상적으로 유의미한 용량에서 관찰되는 독성은 무엇인가? | 안전 한계(safety margins)와 향후 모니터링 항목을 정의합니다. | 일반 독성 자료, 간 효소치 변화, 补체 활성 마커, 후근신경절(DRG) 조직검사 결과, 혈액학적 지표 및 사이토카인 수치. |
| 임상용 제품이 전임상 제품과 비교하여 동등한가? | 투여량 계산 신뢰도의 기초는 제품의 동등한 품질에 기반합니다. | 게놈 역가, 역가(potency) 시험, 빈/가득 찬 비율, 게놈 무결성, 불순물 및 응집 유무 분석. |
| 임상 현장에서 잠재적 위험을 어떻게 통제할 것인가? | 안전성은 초기 용량뿐만 아니라 임상 프로토콜의 안전장치 설계에 의해 결정됩니다. | 센티넬 투여 수립, 시차 환자 등록법, 투여 중단 규칙 설계, 면역억제제 투여 계획 및 장기 추적 조사 계획. |
6. 케이스 스터디: AAV9-SMN1 개발이 남긴 교훈
척수성 근위축증(SMA)은 AAV 투여량 환산의 가장 대표적이고 중요한 이정표를 보여주는 사례입니다. 졸겐스마(Zolgensma, onasemnogene abeparvovec)는 SMA를 앓고 있는 소아 환자에게 기능적인 SMN1 이식유전자를 전달하기 위해 설계된 AAV9 기반 유전자 치료제입니다. 이 승인된 제품은 1회 정맥 주사로 1.1 × 10^14 vg/kg 용량이 투여됩니다.
SMA 프로그램은 왜 마우스 모델에서의 효능 데이터가 투여량 선택의 한 가지 조각에 불과한지 보여줍니다. 신생아 SMA 마우스 모델에서 AAV9-SMN 전달은 강력한 생존율 개선과 운동기능 향상을 보여주었습니다. 하지만 최종 임상 용량은 마우스 투여량을 단순히 vg/kg 기준으로 동일하게 환산하는 것으로 정당화될 수 없었습니다. 개발사들은 추가적으로 체내 분포, 대동물 안전성 프로파일, 간 효소 수치 상승 현상, 후근신경절(DRG) 소견, 환자의 나이, 기저 질환 상태, 스테로이드 병용 관리 기법 및 임상 관찰 프로토콜을 종합적으로 이해해야만 했습니다.
또한 대동물 대상 고용량 전신 투여 AAV 연구를 통해, 마우스 모델에서는 미처 예측하기 어려웠던 독성이 대동물 종과 연구 환경에서 발현될 수 있음이 증명되기도 했습니다. 인간 SMN을 전달하는 고용량 정맥 투여 AAV에 관한 한 연구 논문에서는 비인류 영장류(NHP)와 자돈(새끼 돼지)에서 간 손상과 감각 신경세포 이상을 포함한 중증 독성이 보고된 바 있습니다. 이것이 모든 전신 투여 AAV 프로그램이 반드시 같은 위험을 갖는다는 뜻은 아니지만, 투여량 설계가 단순한 수학 공식이 아닌 ‘수렴하는 여러 과학적 증거(converging evidence)’들로 엮여 완성되어야 함을 일깨워 줍니다.
표 4. 척수성 근위축증(SMA)의 AAV9-SMN1 개발 단계별 교훈
| 개발 단계 | 모델 및 대상군 | 투여량 정보 | 핵심 교훈 |
| 마우스 효능 연구 | 신생아 SMA 마우스 모델 | 초기 투여 시 강력한 생존율 및 운동기능 향상 관찰. | 마우스 효능은 개념 입증(Proof of Concept)을 지지하지만, 인간 임상 용량을 직접 정의할 수는 없음. |
| 대동물 평가 | NHP 및 기타 대동물 모델 | 고용량 전신 투여 연구에서 마우스 연구를 통해 다 예측할 수 없었던 간 손상 및 감각 신경세포 독성 위험 인지. | 대동물 데이터는 전신 투여 AAV의 안전성 평가 시 매우 중대한 요소임. |
| 초기 임상 시험 | 소아 SMA 환자군 | 영유아 SMA 환자를 대상으로 전신 AAV9-SMN1 투여 평가. | 임상 용량 설정은 효능, 생체 내 분포, 독성학, 안전성 모니터링 수단 및 환자의 기저 상태가 긴밀하게 통합되어 결정됨. |
| 최종 승인 임상 적용 | 치료 승인 요건을 갖춘 소아 SMA 환자 | 1회 정맥 주사, 1.1 × 10^14 vg/kg 투여. | 최종 승인 용량은 마우스 투여량의 직접적인 변환이 아닌, 제품에 특화되어 종합 수립된 변환 증거를 기반으로 결정됨. |
7. AAV 투여량 환산 시 흔히 범하는 오류들
많은 투여량 설계 실패 사례는 계산 공식 뒷단에 있는 가정들을 명확히 기술하지 않아 발생합니다. 특히 사전 IND(Pre-IND) 준비 단계에서 다음과 같은 오류들이 흔히 나타납니다.
표 5. AAV 투여량 설계 시 주요 오류와 개선책
| 대표적인 오류 (Pitfall) | 이것이 유발하는 위험 요소 | 바람직한 대안적 접근법 (Better approach) |
| 마우스 vg/kg을 인간 vg/kg으로 단순 1:1 변환하는 것 | 종 간의 장기 크기, 생체 내 분포, 형질도입 효율 및 면역 반응의 생물학적 차이를 완전히 무시하게 됨. | 마우스 데이터를 개념 입증용으로 활용한 후, 표적 장기 기준 스케일링과 대동물 데이터를 통합해 계산하십시오. |
| 역가 정보(Potency context) 없이 총 vg 수치만 사용하는 것 | 게놈 복제수(genome copies) 자체가 벡터가 보여주는 기능적 활성을 항상 정비례로 대변하지는 않음. | 투여량 정보에 역가(potency), 감염성, 게놈 무결성 및 빈/가득 찬 캡시드 비율 분석 데이터를 반드시 연계하십시오. |
| NHP 데이터를 완벽히 예측 가능한 모델로 신뢰하는 것 | 비인류 영장류(NHP) 연구가 우수한 정보를 주지만 인간과 100% 동일하지는 않음. | NHP 데이터를 투여량 이행의 든든한 기준(Anchor)으로 삼되, 불확실성 영역이 남아있음을 명시하십시오. |
| 고용량 독성을 제어하기 위해 오직 면역억제제 투여에만 의존하는 것 | 면역억제제 병용은 면역성 손상을 완화할 뿐, 안전하지 못한 설계 용량 자체를 안전하게 만들어 주지는 못함. | 초기 투여 시작 용량, 증량 설계 계획 및 투여 중단 기준(Stopping criteria)을 원점에서 재평가하십시오. |
| 안구 또는 국소 CNS 전달 경로에 vg/kg 기준을 적용하는 것 | 국소적인 노출 정도는 전신 체중이 아닌 해당 구획의 해부학적 크기와 주입 부피에 지배를 받음. | 안구당(vg/eye) 혹은 주사 부위당(vg/injection site)과 같은 ‘구획 특이적 단위’를 사용하십시오. |
| scAAV가 항상 ssAAV보다 고정된 배수만큼 더 강력할 것이라 단정하는 것 | 벡터의 효능은 조직 타입, 프로모터, 이식유전자, 캡시드 구조 및 동물 모델에 따라 상이함. | 고정 관념에 의존하지 말고, 해당 물질 특화형 비교 역가 데이터를 직접 생성하십시오. |
8. 프로그램 유형별 AAV 투여량 설계 가이드라인
투여량 설정 시의 불확실성을 낮추는 실무적인 방법은 개발하려는 프로그램의 특성에 용량 결정 논리를 맞추는 것입니다. 간, 망막, 중추신경계, 근육 및 소아 대상 프로그램은 저마다 다른 가정과 증거 패키지를 수립해야 합니다.
표 6. 프로그램 유형별 실무 AAV 용량 설정 가이드라인
| 프로그램 타겟 유형 | 권장 용량 설정 논리 | 우선 확보해야 할 데이터 영역 |
| 간 표적 AAV | 표적 장기 중심 스케일링 방식과 NHP 또는 타 적절한 대동물의 데이터 결합을 검토하십시오. | 간 생체 내 분포도, 간세포 형질도입 비율, 분비 단백질 농도, ALT/AST 변화량, 빌리루빈 및 응고계 바이오마커. |
| 안구 투여 AAV | vg/kg 단위를 일체 지양하고, vg/안구 또는 vg/주사 방식을 채택하십시오. | 표적 망막 세포 수, 주입 부피, 국소 면역 반응 정도, 망막부 해부학 구조 및 벡터 확산 범위. |
| 중추신경계(CNS) 투여 AAV | 전달 경로 고유의 해부학적 크기와 체액 내 확산 거동을 토대로 용량을 결정하십시오. | 뇌척수액(CSF) 부피, 표적 영역의 커버리지율, 후근신경절(DRG) 노출도, 국소 내약성 및 생체 내 분포 분석. |
| 전신 근육 유전자 치료 AAV | vg/kg 단위와 함께 체내에 축적되는 총 벡터 부하량(total vg burden)을 동시에 평가하십시오. | 근육 발현 효율, 간 내 흡수율, 补체 활성 유무, 총 환자 체중 및 질환 진행 단계. |
| 소아 투여 AAV | 발달 단계에 있는 소아 생리학적 특성과 장기적 추적 시의 잠재 위험을 반드시 고려하십시오. | 연령별 장기 비율 변화, 면역 체계의 성숙 정도, 발달 성장 속도, 소아 특화 안전성 관찰 데이터 및 장기 추정 분석. |
9. 성공적인 Pre-IND Dose Rationale 작성을 위해 포함해야 할 사항들
잘 준비된 Pre-IND 브리핑 패키지(briefing package)는 투여량 선택 과정을 하나의 통합된 일련의 논증 구조로 보여주어야 합니다. 목적은 완벽한 계산 결과를 입증하는 것이 아니라, 설정한 가정이 과학적으로 타당하고, 불확실성이 존재하는 부분을 충분히 인지하고 있으며, 임상 프로토콜 설계가 잠재적 위험 요소를 안전하게 관리할 수 있음을 입증하는 것입니다.
표 7. Pre-IND AAV 투여량 타당성 보고서의 권장 구성 요소
| 문서 구성 섹션 | 포함되어야 할 핵심 내용 및 데이터 |
| 제안 임상 용량 범위 | 초기 임상 투여량, 증량 단계 용량 설정치, 계획된 최대 허용 한계량 및 설정 배경 근거. |
| 동물 효능 데이터 | 최소 유효 용량 데이터, 용량-반응 곡선 지표 및 임상 평가 질환 모델과의 실질적 부합 수준 기술. |
| 생체 내 분포 (Biodistribution) | 표적 및 표적 외 조직의 노출 수준, 잔류 정도, 클리어런스 프로파일 및 조직별 이식유전자 복제수 정보. |
| 독성 평가 (Toxicology) | 최대무독성용량(NOAEL) 혹은 유해 반응 발현 용량 정보, 유해 관찰 소견, 회복성 유무 및 확보한 안전 한계(margins). |
| 제품의 분석적 특성 | 역가 측정 분석법 종류, 역가 검증 자료, 빈/가득 찬 비율, 게놈 무결성 검사, 함유 불순물 규정 및 동등성 자료. |
| 전달 경로 및 방식 | 특정 투여 경로를 채택한 과학적 사유와 그에 따른 경로 특이적 안전 대책 설명. |
| 대상 환자군 프로파일 | 치료 대상 질환의 진행 상태, 환자 연령 및 체중대 분포, 장기 기능 수준, 사전 치료 이력 및 기존 면역 상태. |
| 임상적 위험 경감 방책 | 센티넬 투여 방식 수립, 환자 등록 주기 조절, 투여 중단 가이드라인 설정, 모니터링 체계 및 동반 면역억제 전략. |
| 불확실성 및 핵심 가정 | 현재 규명된 정보와 추정치로 적용한 정보, 그리고 여전히 과학적 규명이 필요한 영역의 투명한 기록. |
10. 자주 묻는 질문 (FAQ)
Q. vg/동물 단위를 vg/kg으로 변환하려면 어떻게 하나요?
A: 단순 계산은 명확합니다. 총 주입된 벡터 게놈 수(vg)를 해당 동물의 체중(kg)으로 나누면 됩니다. 다만 이 과정에서 선행되어야 할 질문은 “논문 등에서 보고된 역가 수치가 실제 활성을 가진 벡터 게놈을 정확히 반영하는가?”입니다. 문헌 정보를 활용하기 전에 사용된 역가 분석법, 빈/가득 찬 비율 보고 유무, 실제 역가 수준 및 해당 벡터 제조 품질이 귀사에서 임상용으로 생산할 벡터 제조 품질과 비교하여 동등한 수준인지 확인하십시오.
Q. FDA나 EMA가 공식적으로 제공하는 AAV FIH 용량 설정 공식이 존재하나요?
A: 존재하지 않습니다. 규제 기관들은 정형화된 공식의 일률적인 대입보다는 각 프로그램 고유의 과학적 정당성을 갖춘 투여량 근거(rationale) 제출을 요구합니다. FDA 가이던스에서는 가용한 임상 정보, 유사 물질 개발 경험, 동물 데이터, in vitro 자료, 예측 시뮬레이션 모델 및 적절한 수준의 알로메트릭 스케일링 지표 등을 종합적으로 참조해 용량을 결정해야 한다고 명시합니다. EMA 가이드라인 또한 품질 분석 및 비임상 결과를 주축으로 제품 자체의 역가와 밀접히 연결된 타당성이 과학적 자료로 증명되어야 한다고 명시하고 있습니다.
Q. 비인류 영장류(NHP) 데이터 없이도 IND 제출이 가능한가요?
A: 프로그램에 따라 다릅니다. 국소 부위 투여 혹은 위해도가 상대적으로 낮은 일부 로컬 AAV 프로그램의 경우, 과학적 정당성이 탄탄히 뒷받침되고 합리적인 수준의 안전 한계와 위해 관리책이 마련되어 있다면 NHP 데이터 없이도 패키지가 수용되는 사례가 있습니다. 그러나 고용량 전신 투여 AAV 프로그램의 경우, 관련 대동물의 생체 내 분포 및 독성 평가 데이터가 결여되어 있다면 간 독성, 후근신경절(DRG) 독성, 补체 활성 등의 안전성 우려와 맞물려 IND 승인이 크게 지연되거나 불허될 규제적 리스크가 매우 큽니다.
Q. NHP 연구에서 간 수치가 상승하는 양상은 무조건 더 강한 면역억제 치료를 통해서만 통제해야 하나요?
A: 아닙니다. 면역억제제는 유용한 위해 감소 수단이 될 수 있지만, 그 자체만으로 근본적으로 안전하지 않은 투여 용량을 안전하게 정당화하는 방패막이가 될 수는 없습니다. 빌리루빈 수치 변화, 응고 지표 이상, 补체 활성화, 염증성 지표 또는 조직 병리 소견을 동반하는 용량 의존적 간 효소치 상승은 ‘허용 용량 범위(dose window)’를 이탈하고 있다는 중요한 안전성 신호입니다. 이러한 경우 임상 시작 용량 및 용량 증량 계획 자체를 보수적으로 재조정해야 합니다.
Q. 용량 설정 과정에서 scAAV(자가 상보적 AAV)는 어떻게 해석해야 합니까?
A: 자가 상보적(Self-complementary) AAV는 2차 가닥 합성(second-strand synthesis) 단계를 생략하므로 특정 연구 환경에서 단일 가닥 AAV(ssAAV)보다 빠르고 강한 발현 유도 성능을 보여줄 수 있습니다. 그러나 이러한 효과는 제품 사양, 표적 조직 유형, 프로모터 종류, 탑재 유전자 및 적용 대상 생물 모델에 맞춰 고유하게 나타납니다. 직접 수행한 동등성 비교 데이터 없이 단순히 규격화된 몇 배수의 효능 가중치를 일률적으로 적용하는 것은 과학적으로 바람직하지 않습니다.
11. 핵심 요약 (Key Takeaways)
- 원하는 임상 효능 정의가 우선: 동물 모델에서의 유효 용량 수치를 기계적으로 옮기기 전에, 실제 인간 치료에 필요한 생물학적 작용 기준이 무엇인지 명확히 설정하십시오.
- 마우스는 개념 검증용: 마우스 데이터는 작동 메커니즘을 입증하는 유용한 도구이지만, 마우스에서 확인된 vg/kg 수치를 인간에게 그대로 대입하여 투여량을 설정해서는 안 됩니다.
- 경로별 스케일링 논리 적용: 유효량 스케일링 논리를 타겟 조직 특성 및 투여 전달 경로에 맞추어 개별화 설계하십시오.
- 전신 투여 시 다각적 검토: 전신 정맥 투여 AAV의 경우, 단순 체중 비율(vg/kg)뿐만 아니라 체내에 가해지는 총 벡터 캡시드 부하량(total vector burden)을 심각하게 다루어야 합니다.
- 대동물 앵커 활용: 전신 노출 비중과 안전성 위해성이 크다고 판단되는 경우, NHP 혹은 목적에 부합하는 대동물 평가 결과를 신뢰성 있는 전이 기준점(translational anchor)으로 적극 수립하십시오.
- 제품 품질과 역가 통합: 물질 본연의 역가(potency), 완제품 품질 지표 및 전임상-임상 배치 간의 비교 동등성 검토 결과가 용량 타당성 수립 논리에 내재화되어야 합니다.
- 가정의 투명한 기록: Pre-IND 단계 규제 기관과의 밀도 높은 미팅에 앞서, 적용한 모든 가설적 수치와 가정들의 배경 원천을 서류에 투명하게 기록해 두십시오.
많은 유전자 치료제 IND 제출 지연 사례들은 ‘완벽한 스케일링 공식의 부재’ 때문이 아니라, 불명확한 수치 대입 가정, 배치 간 불완전한 품질 동등성, 취약한 역가 증명 논리, 혹은 동물 연구 결과와 인체 생물학 간의 무리한 비약 때문에 발생합니다. 규제 미팅에 돌입하기 전 용량 설정 근거를 간결한 내부 타당성 보고서 형식으로 작성해 보는 것은 개발 담당자, CRO 파트너, 독성 전문가, CMC 책임자 및 규제 자문위원단이 동일한 정량적 가정하에 일목요연하게 방향성을 맞출 수 있는 훌륭한 전략입니다.
PackGene의 AAV 투여량 환산 및 IND 지원 역량
AAV 투여량 설계는 복잡한 생물학적 이해뿐만 아니라 벡터의 품질 유지력, 역가 관리 역량 및 제조 공정의 일관성과도 직결됩니다. 팩진(PackGene)은 초기 후보 물질 발굴 단계부터 IND-enabling 전임상 단계, 그리고 GMP 완제품 생산에 이르기까지 벡터 디자인, 플라스미드 최적화, AAV 패키징 서비스, 제조 공정 개발, 정밀 분석 분석법 확립 및 규제 대응 문서 지원에 이르기까지 원스톱 솔루션을 제공합니다.
Pre-IND 단계와 임상 진입을 앞둔 연구팀의 경우, 초기 설계 단계부터 투여량 스케일링 전략과 공정 개발 전략을 유기적으로 연계해야 시행착오를 줄일 수 있습니다. 고품질 벡터 생산 역량, 신뢰도 높은 역가 측정 기술, 불순물 통제력, 생체 내 분포 연구 노하우 및 배치 간 분석 동등성 보고서 확보는 FIH 투여량을 성공적으로 정당화하고 규제 기관의 검증을 통과하는 가장 든든한 초석이 될 것입니다.
- 연구용, 비임상 전임상용 및 GMP 수준의 AAV 맞춤 생산 지원
- AAV 벡터 디자인 및 플라스미드 구조 최적화 기술
- 균일한 고품질 벡터 유지를 위한 제조 공정(PD) 고도화 및 스케일업 솔루션
- 벡터 역가, 순도, 게놈 무결성, 빈/가득 찬 비율 및 불순물 수준을 검증하는 정밀 분석 시스템 구축
- IND 제출 자료 구축을 지원하는 체계적인 CMC 문서 작성 지원
AAV 투여량 환산은 결국 정밀한 생물학적 증거에 기반해 잠재 위험 요소를 합리적으로 관리해 나가는 고도의 의사결정 과정입니다. 초기 개발 설계에서 투여량 스케일링 로드맵을 완벽하게 수립하는 것은 개발 과정에서의 막대한 위험 요소를 경감시키고, 소중한 유전자 치료제 프로그램을 동물 시험 단계를 넘어 임상 치료 현장으로 인도하는 가장 빠르고 정확한 지름길이 될 것입니다.
참고문헌
U.S. Food and Drug Administration. Human Gene Therapy for Rare Diseases: Guidance for Industry. January 2020.
European Medicines Agency. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. EMA/CAT/80183/2014.
Burr A, Erickson P, Bento R, Shama K, Roth C, Parekkadan B. Allometric-like scaling of AAV gene therapy for systemic protein delivery. Mol Ther Methods Clin Dev. 2022;27:368-379. doi:10.1016/j.omtm.2022.10.011.
Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285-298. doi:10.1089/hum.2018.015.
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713-1722. doi:10.1056/NEJMoa1706198.
Zolgensma (onasemnogene abeparvovec-xioi) prescribing information. Novartis Gene Therapies. https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf
Francois A, Bouzelha M, Lecomte E, et al. Accurate titration of infectious AAV particles requires measurement of biologically active vector genomes and suitable controls. Mol Ther Methods Clin Dev. 2018;10:223-236. doi:10.1016/j.omtm.2018.07.004.
About PackGene
PackGene Biotech is a world-leading CRO and CDMO, excelling in AAV vectors, mRNA, plasmid DNA, and lentiviral vector solutions. Our comprehensive offerings span from vector design and construction to AAV, lentivirus, and mRNA services. With a sharp focus on early-stage drug discovery, preclinical development, and cell and gene therapy trials, we deliver cost-effective, dependable, and scalable production solutions. Leveraging our groundbreaking π-alpha 293 AAV high-yield platform, we amplify AAV production by up to 10-fold, yielding up to 1e+17vg per batch to meet diverse commercial and clinical project needs. Moreover, our tailored mRNA and LNP products and services cater to every stage of drug and vaccine development, from research to GMP production, providing a seamless, end-to-end solution.

Related Services
-
AAV QC 분석 서비스
-
유전자 서열의 정확성, 순도, 효능, 안전성에 대한 전면적인 QC 분석 제공
- 규제 요건에 부합하는 맞춤형 검증 분석법 지원
- GMP 등급의 품질 관리 및 제품 출고 지원으로 신속한 납기 보장

-
-
AAV Packaging – Research Grade
-
고순도, 저엔도톡신, 낮은 empty 캡시드 비율의 품질 보장
-
70종 이상의 혈청형 및 50,000건 이상의 프로젝트 수행 경험
-
전문가의 맞춤형 기술 지원으로 신속하고 신뢰할 수 있는 서비스 제공
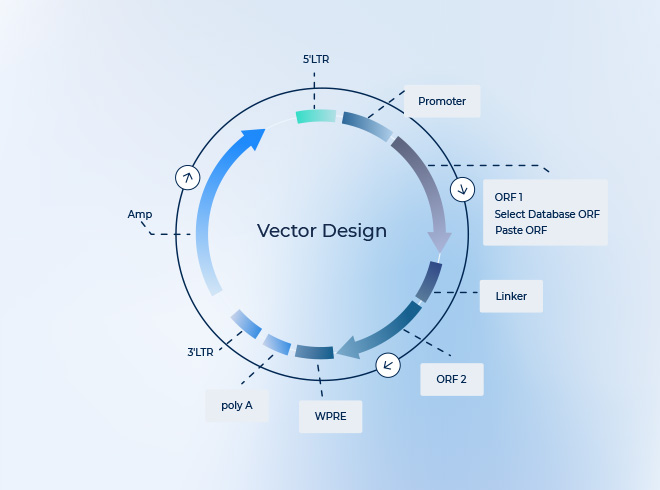
-
-
AAV Packaging – NHP Grade
- 매우 낮은 엔도톡신 철저한 오염 관리 시스템
- ddPCR 기반 정밀 titer 측정 및 유전체 완전성 확인 완료
- 일관된 결과로 향상된 안전성 및 유효성 보장
-
AAV 제품
- 즉시 사용 가능한 제품, 신속한 출고 가능
- 다양한 연구 목적을 아우르는 폭넓은 제품 라인업
- 엄격한 품질 관리(QC)로 신뢰할 수 있는 품질 보장
-
AAV 플라스미드 디자인 및 제작
- 유전자 전달 효율을 극대화하는 맞춤형 AAV 플라스미드 제작
- piVector 플랫폼을 통한 용도별 요소 선택과 간편한 설계 지원
- CRISPR, shRNA 및 다양한 AAV 벡터 유형에 폭넓게 대응