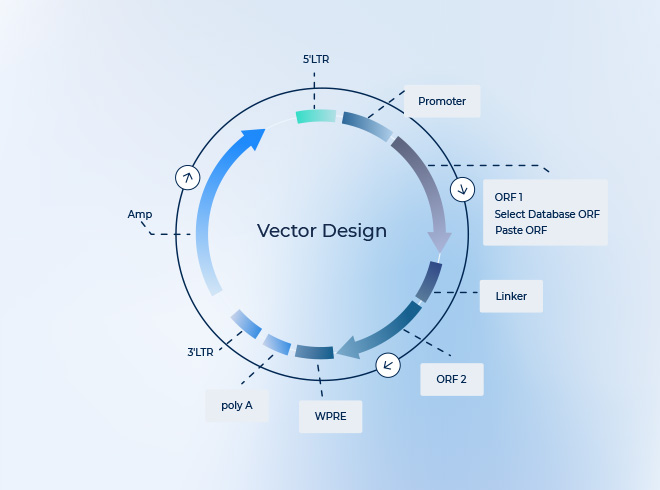
서론
더 많은 유전자치료제 개발 프로그램이 임상시험계획(IND) 신청 단계로 진전됨에 따라, 아데노연관바이러스(adeno-associated virus, AAV) 제품에 대한 CMC(Chemistry, Manufacturing, and Controls; 품질·제조·관리) 관련 기대 수준은 점점 더 엄격해지고 있습니다. 초기 탐색 단계에서는 많은 팀이 주로 벡터 게놈 역가(vector genome titer)가 충분히 높은지에 집중합니다. qPCR 또는 ddPCR을 통해 vg/mL로 보고되는 깔끔한 결과는 해당 프로그램이 다음 단계로 나아갈 준비가 되었다는 확신을 줄 수 있습니다.
그러나 AAV 제품에 대한 규제 검토는 역가에서 멈추는 경우가 거의 없습니다. 심사자는 의뢰자가 제품의 핵심 품질 특성(Critical Quality Attributes, CQA)을 이해하고 관리하고 있는지 확인하려는 경우가 더 많습니다. 특히 중요한 질문은 다음 두 가지입니다.
- 빈 캡시드 대비 충전 캡시드의 비율(empty-to-full capsid ratio)은 얼마인가?
- 제조 배치 간 결과는 얼마나 일관적인가?
이러한 질문은 AAV 프로그램이 IND 지원 개발 단계에 진입할 준비가 되었는지, 또는 추가적인 공정 개발, 분석적 특성 규명 및 비교동등성 평가가 필요한지를 결정할 수 있습니다.
높은 벡터 게놈 역가는 출발점이 될 수는 있지만, 제품 품질을 입증하기에는 충분하지 않습니다. AAV CMC에서 배치 간 일관성과 캡시드 품질은 단일한 매력적인 vg/mL 수치보다 더 중요한 경우가 많습니다.
AAV 역가의 한계
사람들이 AAV 역가를 언급할 때 일반적으로 의미하는 것은 qPCR 또는 ddPCR을 사용해 mL당 벡터 게놈 수로 측정되는 벡터 게놈 역가입니다. 이 값은 시료 내에서 검출 가능한 표적 게놈 사본 수를 나타냅니다. 이는 필수적인 측정값이지만, 전체 이야기를 설명해 주지는 않습니다.
AAV 제조 공정에서는 다음을 포함한 이질적인 입자 집단이 생성됩니다.
- 의도한 치료용 게놈을 포함하는 충전 캡시드(full capsids)
- 단백질 외피는 있지만 벡터 게놈이 없는 빈 캡시드(empty capsids)
- 불완전한 게놈 또는 핵산 조각을 포함하는 부분 포장 캡시드(partially packaged capsids)
- 의도하지 않았거나 표적이 아닌 핵산 종을 포함할 수 있는 비정상 포장 캡시드(aberrantly packaged capsids)
qPCR과 ddPCR은 특정 벡터 게놈 서열을 정량합니다. 이들 방법은 총 캡시드 수, 빈 캡시드, 캡시드 단백질 함량 또는 포장된 게놈이 온전하고 기능적인지 여부를 직접 측정하지 않습니다. 따라서 동일한 vg/mL 값을 가진 두 개의 AAV 로트라도 효능, 안전성 프로파일, 불순물 부담 및 임상적 적합성 측면에서 상당히 다를 수 있습니다.
실무적으로 볼 때, 벡터 게놈 역가는 유효 용량과 동일하지 않습니다. 높은 역가라 하더라도 기능하지 않거나 충분히 특성 규명되지 않은 입자의 비율이 높을 수 있습니다.
빈 캡시드 비율이 중요한 이유
빈 캡시드는 의도한 유전적 탑재물을 운반하지 않기 때문에 치료 활성이 없습니다. 그러나 생물학적으로 무의미한 것은 아닙니다. 빈 캡시드는 여전히 세포와 상호작용하고, 수용체에 결합하며, 총 캡시드 부담에 기여하고, 면역 반응에 영향을 미칠 수 있습니다.
생산 시스템, 플라스미드 비율, 수확 조건 및 정제 전략에 따라 빈 캡시드와 부분 충전 캡시드는 최종 AAV 제제의 상당한 비율을 차지할 수 있습니다. 따라서 빈 캡시드 비율은 부차적인 분석 세부사항이 아니라 핵심 품질 특성으로 점점 더 인식되고 있습니다.
전신 투여용 AAV 제품의 경우, 환자가 높은 총 캡시드 부하에 노출될 수 있기 때문에 이 문제는 더욱 중요해집니다. 빈 캡시드 비율이 높은 제형은 치료용 게놈 전달의 상응하는 증가 없이 환자를 더 많은 캡시드 단백질에 노출시킬 수 있습니다.
빈 캡시드 특성 규명을 위한 분석 방법
빈 캡시드 데이터의 신뢰성은 사용된 분석 방법에 크게 좌우됩니다. 완벽한 단일 방법은 없으며, 적절한 전략은 개발 단계, 시료 가용성, 규제 기대치 및 제품별 위험에 따라 달라집니다.
분석 초원심분리법
분석 초원심분리법(Analytical Ultracentrifugation, AUC)은 침강 거동을 기반으로 입자를 분리합니다. AUC는 빈 캡시드, 충전 캡시드 및 부분 충전 캡시드 집단을 구별할 수 있으며, AAV 캡시드 특성 규명을 위한 강력한 기준 방법으로 널리 인정받고 있습니다.
AUC는 정량적이고 비교적 신뢰도 높은 정보를 제공하지만, 시간이 많이 걸리고 처리량이 낮으며 전문적인 지식과 장비가 필요합니다. 또한 일반적으로 일부 신흥 방법보다 더 많은 시료가 필요합니다. 이러한 한계에도 불구하고 AUC는 GMP 출하 시험, 비교동등성 평가 및 규제 제출 패키지에서 가장 중요한 도구 중 하나로 남아 있습니다.
SEC-MALS
다각도 광산란 검출기와 결합한 크기배제 크로마토그래피(Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering, SEC-MALS)는 AAV 입자를 다른 종으로부터 분리하고 분자량 및 크기 관련 특성을 측정합니다. 이 방법은 캡시드 내용물, 응집, 순도 및 입자 크기 분포에 관한 유용한 정보를 제공할 수 있습니다.
SEC-MALS는 AUC보다 빠르고 운영상 더 편리하므로 공정 개발에 유용하며, 적절한 방법 적격성 평가 또는 밸리데이션 이후에는 출하 시험에도 잠재적으로 활용될 수 있습니다. 다만 복잡한 충전, 빈 및 부분 충전 캡시드 집단에 대한 분해능은 AUC보다 낮을 수 있습니다.
미세유체 저항성 펄스 센싱
Samux-MP와 같은 미세유체 저항성 펄스 센싱 플랫폼은 입자가 나노스케일 기공을 통과할 때 이를 특성 규명합니다. 통과 중 생성되는 신호는 입자 크기 및 캡시드 충전 상태와 관련된 정보를 제공할 수 있습니다.
이러한 방법은 빠르고 소량의 시료만 필요할 수 있습니다. 공정 개발과 고도 특성 규명에 유용하지만, 더 넓은 규제 수용성은 제품별 밸리데이션, 방법의 견고성, 그리고 확립된 직교 분석법과의 상관관계 입증에 달려 있습니다.
투과전자현미경
투과전자현미경(Transmission Electron Microscopy, TEM)은 AAV 입자를 직접 시각화할 수 있게 하며, 입자 형태와 겉보기 캡시드 충전 상태에 대한 직관적인 근거를 제공할 수 있습니다.
TEM은 초기 공정 개발 단계에서 유용하지만 한계가 있습니다. 계수되는 입자 수가 제한적인 경우가 많고, 해석이 작업자 의존적일 수 있으며, 정밀한 정량 출하 시험에는 이상적이지 않습니다. TEM은 일반적으로 빈 캡시드 관리의 유일한 근거가 아니라 보조적 또는 직교적 근거로 사용되어야 합니다.
A260/A280 비율
A260/A280 흡광도 비율은 핵산 및 단백질 함량을 추정하며, 빠르고 저비용의 근사치를 제공할 수 있습니다. 그러나 이 비율은 단백질 불순물, 핵산 조각, 첨가제, 완충액 조성 및 pH의 영향을 크게 받습니다.
AAV 제품의 경우, A260/A280만으로는 규제 수준의 빈 캡시드 특성 규명에 충분하지 않습니다. 내부 공정 모니터링에는 유용할 수 있지만, IND 지원 또는 GMP 단계 개발을 위한 포괄적인 품질 평가로 제시되어서는 안 됩니다.
빈 캡시드에 대한 규제 관점
규제기관은 유전자치료제에 대해 의뢰자가 제품 관련 및 공정 관련 불순물을 이해하고, 특성 규명하며, 관리할 것을 기대합니다. AAV 벡터의 경우 여기에는 캡시드 집단, 벡터 게놈 무결성, 효능, 잔류 숙주세포 DNA, 잔류 플라스미드 DNA, 숙주세포 단백질, 응집체 및 기타 제품 또는 공정 관련 불순물의 특성 규명이 포함됩니다.
현재 모든 AAV 제품에 적용되는 빈 캡시드에 대한 보편적인 규제 출하 기준은 없습니다. 대신 허용 가능한 수준은 제품, 투여 경로, 용량, 적응증, 환자 집단, 비임상 안전성 패키지 및 임상적 위험-편익 프로파일을 기반으로 정당화되어야 합니다.
Dark Horse Consulting의 업계 제안 초안은 빈 AAV 캡시드에 대한 최대 출하 기준을 제안한 바 있으나, 이 제안은 FDA에 의해 구속력 있는 규제 요건으로 공식 채택되지 않았습니다. 따라서 빈 캡시드 관리는 규제상 기대사항이며, 구체적인 허용 기준은 제품별로 설정되고 과학적으로 정당화되어야 한다고 표현하는 것이 더 정확합니다.
배치 간 일관성은 핵심 CMC 요건
역가와 빈 캡시드 비율은 중요하지만, 재현 가능하지 않다면 충분하지 않습니다. 배치 간 일관성은 제조 공정이 관리되고 있으며 비교 가능한 품질 특성을 가진 물질을 생산할 수 있음을 입증합니다.
AAV 제품의 경우, 배치 일관성 평가는 일반적으로 다음 항목을 포함해야 합니다.
- 벡터 게놈 역가
- 총 캡시드 농도
- 빈, 부분 충전 및 충전 캡시드의 분포
- 게놈 무결성
- 효능 또는 생물학적 활성
- 순도 및 불순물 프로파일
- 잔류 숙주세포 DNA 및 숙주세胞 단백질
- 잔류 플라스미드 DNA
- 응집
- 해당되는 경우 무균성, 엔도톡신 및 생물부하
- 안정성 지표 특성
단일 고성능 배치만으로는 이후 배치에서 큰 변동성이 나타날 경우 설득력이 떨어질 수 있습니다. CMC 관점에서는 일관되게 재현할 수 없는 높은 역가보다, 다소 낮더라도 재현 가능한 역가가 더 가치 있을 수 있습니다.
예를 들어, 조절된 빈 캡시드 수준과 함께 중등도 역가의 AAV를 반복적으로 생산하는 공정은, 때때로 매우 높은 역가를 생산하지만 캡시드 품질이 크게 변동하는 공정보다 임상적 및 규제적 측면에서 더 의미 있을 수 있습니다.
예시적 산업 시나리오
1 × 10¹³ vg/mL로 보고된 로트를 사용하여 동물 효능시험을 완료한 AAV 프로그램을 생각해 보겠습니다. 결과는 우수해 보이고, 팀은 IND 지원 활동 준비를 시작합니다.
제출 전, 의뢰자는 AUC 시험을 의뢰하고 해당 로트에 높은 비율의 빈 캡시드가 포함되어 있음을 발견합니다. 이 발견은 비임상 물질에 대한 해석을 바꿉니다. 벡터 게놈 역가는 정확했지만, 동물에게 투여된 총 캡시드 부담은 원래 이해했던 것보다 훨씬 높았을 수 있습니다. 또한 제품에는 가정했던 것보다 치료적으로 의미 있는 충전 입자가 더 적게 포함되어 있을 수 있습니다.
이제 팀은 여러 CMC 질문에 직면합니다.
- 관찰된 효능은 의도한 충전 캡시드 집단에 의해 유도되었는가?
- 안전성 프로파일은 높은 총 캡시드 부하의 영향을 받았는가?
- 정제 공정을 개선하여 충전 캡시드를 농축할 수 있는가?
- 개선된 공정이 비교 가능한 효능과 안전성을 재현할 수 있는가?
- 추가적인 브리징 또는 비교동등성 평가가 필요한가?
이러한 시나리오는 역가만으로는 오해를 불러일으킬 수 있음을 보여줍니다. 문제는 역가 측정법이 반드시 “거짓말을 했다”는 것이 아닙니다. 문제는 팀이 불완전한 질문을 했다는 데 있습니다.
더 나은 질문은 단순히 “vg/mL가 얼마인가?”가 아닙니다.
더 나은 질문은 “그 역가 중 어느 정도가 재현 가능하고, 온전하며, 치료적으로 의미 있는 AAV 제품을 나타내는가?”입니다.
AAV 개발자를 위한 실무 지침
탐색 및 초기 연구 단계
초기 연구 단계에서는 qPCR 또는 ddPCR 역가 데이터가 스크리닝 목적으로 허용될 수 있습니다. 그러나 팀은 역가를 품질의 증거로 과도하게 해석하지 않도록 해야 합니다.
권장 조치는 다음과 같습니다.
- 가능한 경우 TEM 또는 다른 직교적 입자 평가를 요청
- 공급업체가 총 입자 데이터를 제공하는지, 아니면 vg/mL만 제공하는지 기록
- 향후 캡시드 품질 조정을 고려할 수 있도록 충분한 유연성을 가진 용량 범위 탐색 연구 설계
- 향후 비교동등성 분석을 위한 기준 물질 보존
Pre-IND 단계
12개월 이내 IND 제출이 예상되는 경우, AAV CMC 작업은 조기에 시작해야 합니다. 분석적 특성 규명을 최종 GMP 배치까지 미뤄서는 안 됩니다.
권장 조치는 다음과 같습니다.
- 벡터 게놈 역가 측정을 위한 ddPCR 또는 과학적으로 충분히 정당화된 qPCR 방법 확립
- 캡시드 특성 규명을 위한 AUC, SEC-MALS 또는 기타 직교 방법 도입
- 게놈 무결성 및 포장된 DNA 종 평가
- 효능 및 벡터 게놈 역가와의 관계 평가
- 공정 변동성을 이해하기 위한 다중 배치 데이터 생성
- 예비 핵심 품질 특성 및 허용 기준 정의
GMP 제조 단계
GMP 단계에서는 제품 위험 및 개발 단계에 따라 빈 캡시드 비율을 제품 규격 또는 관리 전략에 포함하는 것을 고려해야 합니다. 이를 정보 제공용으로만 취급한다면, 의뢰자는 강력한 과학적 근거를 갖추어야 합니다.
권장 조치는 다음과 같습니다.
- 개발 단계에 적합한 적격성 평가 또는 밸리데이션된 분석 방법 사용
- 사전에 정의된 허용 기준 또는 경고/조치 한계 설정
- 배치 간 일관성 입증
- 캡시드 품질을 효능, 안전성 및 용량 설정 근거와 연결
- 규제 검토에 적합한 원자료 및 방법 문서 유지
공급업체 평가
공급업체가 AAV 제품의 “빈 캡시드가 낮다”고 진술하는 경우, 이는 데이터로 뒷받침되어야 합니다. 의뢰자는 다음을 요청해야 합니다.
- 해당되는 경우 원시 AUC 또는 SEC-MALS 데이터
- 방법 적격성 평가 또는 밸리데이션 요약
- 대표 전기영동도, 침강 프로파일 또는 크로마토그램
- 배치 이력 및 비교동등성 데이터
- 충전, 부분 충전 및 빈 캡시드 보고에 대한 명확한 정의
A260/A280에만 의존하는 보고서는 IND 지원 개발을 위한 완전한 AAV 캡시드 품질 패키지로 간주되어서는 안 됩니다.
자주 묻는 질문
Q1: qPCR과 ddPCR이 서로 다른 AAV 역가를 제공합니다. 어느 것을 사용해야 합니까?
qPCR과 ddPCR은 모두 AAV 벡터 게놈 정량에 흔히 사용됩니다. ddPCR은 절대 정량을 제공하고 표준곡선에 의존하지 않는다는 장점이 있습니다. 이는 재현성을 향상시키고 일부 분석 변동성의 원인을 줄일 수 있습니다.
그러나 ddPCR이 모든 경우에 자동으로 옳은 것은 아닙니다. 분석 설계, 프라이머-프로브 위치, 게놈 구조, 시료 준비, 뉴클레아제 처리 및 기준 물질은 모두 결과에 영향을 미칠 수 있습니다. 프로그램이 qPCR에서 ddPCR로 전환하는 경우, 의뢰자는 브리징 연구를 수행하고 규제 제출 자료에서 역가 변화의 원인을 설명해야 합니다.
Q2: 높은 빈 캡시드 비율을 용량 증가로 보상할 수 있습니까?
이는 일반적으로 임상 개발에 적절한 전략이 아닙니다. 용량 증가는 면역 활성화 위험, 간 부담 및 표적 외 안전성 우려를 증가시킬 수 있습니다.
용량 증량은 캡시드 품질의 적절한 관리를 대체할 수 없습니다. 더 나은 접근법은 생산 및 정제 공정을 개선하고, 충분히 특성 규명된 물질을 기반으로 과학적으로 정당화된 용량을 설정하는 것입니다.
Q3: 연구 등급 배치 일관성 데이터가 GMP 배치 데이터를 대체할 수 있습니까?
연구 등급 데이터는 공정 이해를 뒷받침할 수 있지만, 일반적으로 GMP 관련 출하 및 특성 규명 데이터를 대체할 수는 없습니다. 자원이 제한된 경우, 의뢰자는 공정 개발 및 위험 평가를 지원하기 위해 엔지니어링 런을 활용한 후, 임상 사용을 뒷받침하기 위해 GMP 배치 데이터를 사용할 수 있습니다.
최종 전략은 특히 희귀질환 프로그램, 중증 적응증, 가속화된 개발 일정 또는 제조 이력이 제한적인 제품의 경우 규제기관과 논의해야 합니다.
결론
AAV 개발에서 벡터 게놈 역가는 여전히 중요한 측정값이지만, 제품 품질의 유일한 지표로 취급되어서는 안 됩니다. AAV 제품은 복잡한 생물학적 시스템이며, 임상 성능은 vg/mL 이상의 요소에 따라 달라집니다.
핵심 CMC 질문은 다음과 같습니다.
- 제품 중 어느 정도가 충전되어 있고, 온전하며, 치료적으로 의미 있는가?
- 빈 캡시드 또는 부분 충전 캡시드 물질은 얼마나 존재하는가?
- 공정은 배치 간 얼마나 일관적인가?
- 분석 방법은 규제 검토를 뒷받침할 만큼 충분히 견고한가?
- 관리 전략은 제품별 위험과 일치하는가?
높은 AAV 역가는 인상적으로 보일 수 있지만, 제품이 실제로 임상 개발 준비가 되었는지를 결정하는 것은 배치 일관성과 캡시드 품질입니다.
현대 AAV CMC에서 가장 중요한 질문은 더 이상 단순히 “역가가 얼마나 높은가?”가 아닙니다.
그 질문은 “그 역가 중 어느 정도가 일관되고, 기능적이며, 임상적으로 의미 있는 AAV 제품을 나타내는가?”입니다.
참고문헌
Park S, Shin S, Lee H, Jang JH, Lee GM. Enhancing the production of adeno-associated virus (AAV)2 and AAV9 with high full capsid ratio in HEK293 cells through design-of-experiment optimization of triple plasmid ratio. Biotechnology Journal. 2024;19(3).
U.S. Food and Drug Administration. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry.
European Medicines Agency. Guideline on the Quality, Non-clinical and Clinical Aspects of Gene Therapy Medicinal Products.
European Medicines Agency. Reflection Paper on Quality, Non-clinical and Clinical Issues Related to the Development of Recombinant Adeno-Associated Viral Vectors.
Dark Horse Consulting Group. Proposed Draft Guidance for FDA Consideration: Testing of Adeno-Associated Viral (AAV) Vector-Based Human Gene Therapy Products for Empty Capsids During Product Manufacture.
Lock M, Alvira MR, Chen SJ, Wilson JM. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Human Gene Therapy Methods. 2014;25(2):115–125.
About PackGene
PackGene Biotech is a world-leading CRO and CDMO, excelling in AAV vectors, mRNA, plasmid DNA, and lentiviral vector solutions. Our comprehensive offerings span from vector design and construction to AAV, lentivirus, and mRNA services. With a sharp focus on early-stage drug discovery, preclinical development, and cell and gene therapy trials, we deliver cost-effective, dependable, and scalable production solutions. Leveraging our groundbreaking π-alpha 293 AAV high-yield platform, we amplify AAV production by up to 10-fold, yielding up to 1e+17vg per batch to meet diverse commercial and clinical project needs. Moreover, our tailored mRNA and LNP products and services cater to every stage of drug and vaccine development, from research to GMP production, providing a seamless, end-to-end solution.

Related Services
-
AAV QC 분석 서비스
-
유전자 서열의 정확성, 순도, 효능, 안전성에 대한 전면적인 QC 분석 제공
- 규제 요건에 부합하는 맞춤형 검증 분석법 지원
- GMP 등급의 품질 관리 및 제품 출고 지원으로 신속한 납기 보장

-
-
AAV Packaging – Research Grade
-
고순도, 저엔도톡신, 낮은 empty 캡시드 비율의 품질 보장
-
70종 이상의 혈청형 및 50,000건 이상의 프로젝트 수행 경험
-
전문가의 맞춤형 기술 지원으로 신속하고 신뢰할 수 있는 서비스 제공
-
-
AAV Packaging – NHP Grade
- 매우 낮은 엔도톡신 철저한 오염 관리 시스템
- ddPCR 기반 정밀 titer 측정 및 유전체 완전성 확인 완료
- 일관된 결과로 향상된 안전성 및 유효성 보장
-
AAV 제품
- 즉시 사용 가능한 제품, 신속한 출고 가능
- 다양한 연구 목적을 아우르는 폭넓은 제품 라인업
- 엄격한 품질 관리(QC)로 신뢰할 수 있는 품질 보장
-
AAV 플라스미드 디자인 및 제작
- 유전자 전달 효율을 극대화하는 맞춤형 AAV 플라스미드 제작
- piVector 플랫폼을 통한 용도별 요소 선택과 간편한 설계 지원
- CRISPR, shRNA 및 다양한 AAV 벡터 유형에 폭넓게 대응